Оценка нескольких классификаторов, как по отдельности, так и в комбинации, подтверждает решение сосредоточиться на паттернах метилирования внеклеточной ДНК для выявления рака и прогнозирования происхождения ракового сигнала, говорится в презентации на конгрессе ESMO 2021.
"Кроме того, анализ парных образцов тканей и крови участников с раком позволил определить циркулирующую фракцию опухолевых аллелей (cTAF), что дает информацию о клиническом пределе обнаружения и позволяет оценить эффективность обнаружения в сравнении с поведением внеклеточной ДНК", - рассказала Healio Минетта К. Лю, профессор и научный руководитель отделения онкологии и консультант отделения лабораторной медицины и патологии клиники Майо в Рочестере, штат Миннесота.
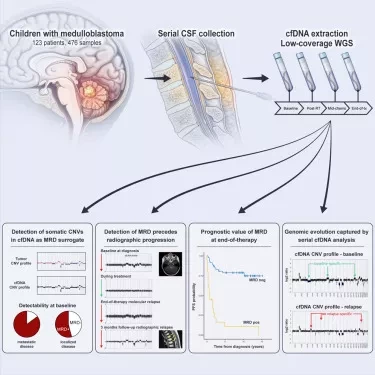
Лю представил результаты первого обсервационного исследования случай-контроль Circulating Cell-free Genome Atlas (CCGA), предназначенного для разработки и валидации теста раннего выявления рака на основе внеклеточной ДНК. Лю и коллеги продолжили исследование после того, как выявили явную необходимость в улучшении и расширении подходов к скринингу, учитывая, что парадигмы популяционного скрининга рака ограничены раком груди, шейки матки, толстой кишки, легких и простаты, и что обнаружение циркулирующей опухолевой ДНК (ctDNA) в периферической крови "естественно подходит" для раннего выявления множества видов рака. "Циркулирующие биомаркеры рака исторически ограничивались профилированием опухоли для подбора лекарственных препаратов при злокачественных опухолях", - отмечает Лю.
"Но последние технологические и научные достижения повысили чувствительность обнаружения ctDNA до такой степени, что молекулярный мониторинг резидуальных заболеваний и скрининг рака по крови стали реальностью".
Анализ включал 2 261 участника: 1414 в независимом обучающем массиве (рак, n = 854; нераковые заболевания, n = 560) и 847 в валидационном массиве (рак, n = 485; нераковые заболевания, n = 362).
Лю и сотрудники собрали и секвенировали плазму и соответствующие лейкоциты крови участников, а также, при наличии, секвенировали биоптаты опухоли. Исследователи использовали данные о полногеномном метилировании, полученные с помощью полногеномного бисульфитного секвенирования, данные о малых соматических вариантах, полученные с помощью целевого секвенирования с коррекцией ошибок, и данные о соматических изменениях числа копий, длине фрагмента, конечной точке фрагмента и аллельном дисбалансе, полученные с помощью полногеномного секвенирования.
После разделения образцов на обучающие и валидационные массивы исследователи обучили 10 классификаторов для выявления солидного рака (карциномы, саркомы и лимфомы). Затем они оценили классификаторы на предмет обнаружения рака и клинического предела обнаружения, который оценивается как вероятность обнаружения рака в зависимости от доли циркулирующих опухолей при использовании соответствующих биоптатов опухолей. Используя полногеномное метилирование, целевое секвенирование и соматические нарушения числа копий, исследователи обучили три дополнительных классификатора для прогнозирования происхождения ракового сигнала и оценки точности.
Результаты показали, что полногеномное метилирование предсказывало происхождение ракового сигнала с точностью 75% и, таким образом, было значительно точнее, чем классификаторы однонуклеотидных вариантов лейкоцитов (35%) или соматических изменений числа копий (41%). Кроме того, клинический предел обнаружения при использовании полногеномного метилирования был более чем в 1,5 раза ниже, чем у любого классификатора полногеномного секвенирования или целевого секвенирования. Предсказание происхождения ракового сигнала было более чем в 1,8 раза более точным при использовании полногеномного метилирования по сравнению с целевым секвенированием или соматическими изменениями числа копий, пишут Лю и коллеги.
Другие основные моменты полногеномного метилирования, сказала Лю во время своей презентации, заключаются в том, что оно не требует удаления биологического фона из парных лейкоцитов, в отличие от однонуклеотидного варианта-лейкоцитов и функции pan, и что оно имеет значительный потенциал для дальнейшего улучшения в достижении более высокой производительности с помощью целевого подхода к анализу метилирования. "Эти результаты отражают текущую работу, направленную на улучшение нашего понимания биологии, связанной с обнаружением ctDNA", - отметила Лю.
Лю и коллеги пришли к выводу, что полногеномное метилирование превосходит другие тесты, не требуя дополнительного секвенирования лейкоцитов для выявления клонального кроветворения, а полученные данные позволяют разработать "значительно улучшенный" тест на целевое метилирование и раннее выявление множественных видов рака для дальнейших исследований в поддержку клинического использования.
"Клинический предел обнаружения является полезным эталоном, по которому можно оценить эффективность тестов на основе внеклеточной ДНК", - сказал Лю в презентации. " Полногеномное метилирование обеспечивает наиболее перспективный подход к внеклеточной ДНК для теста раннего выявления рака на основе анализа крови".